
AID_Project
Member
 |
Nasim Begum | Program-Specific Associate Professor | |
|---|---|---|---|
 |
Maki Kobayashi | Program-Specific Associate Professor | |
 |
I Ketut Gunarta | postdoctoral fellow | |
|
Wencong Wang | doctoral course student | |
 |
Chen Xi | doctoral course student | |
|
Mikiyo Nakata | research assistant | |
|
Maki Sasanuma | research assistant | |
|
Sumiko Matsui | research assistant | |
|
Tabassum Ara | research assistant | |
|
Maidaiti Baerlike | research assistant |
After the completion of mass sequencing project, it appeared that the human genome contains only 20,000-30,000 genes. How can multicellular organisms generate highly sophisticated systems from the limited number of gene?
One of the answers is found in the B lymphocyte system.
B lymphocytes generate virtually unlimited number of diversity of immunoglobulin by its gene alterations. Class switch is a process to diversify the effector function
of the immunoglobulin by conversion of the heavy chain from Cm to Cg, Ca or Ce chain. Class switch has been shown to be mediated by recombination between S regions
located 5' to each CH gene. This group has made important contributions in elucidation of the molecular mechanism for class switch recombination (CSR) and its
regulation: (a) proposal of the deletion model 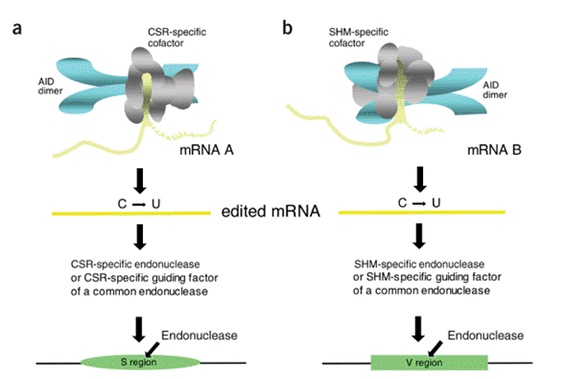 in 1978, (b) identification and characterization of S region as target of CSR in 1980, (c) molecular cloning
and physical mapping of the constant region genes, confirming their order on mouse chromosome predicted by the deletion model in 1982, (d) cloning of IL-4 and
IL-5 involved in regulation of CSR in 1986, and (e) demonstration of circular DNA byproducts excised during CSR in 1990. In 1986, Stavenezer and Alt found that
cytokine stimulation induces transcription from specific intronic promoters located 5' to each S region (germline transcription) and proposed that such transcription
makes recombination target S region accessible to a putative CSR recombinase (accessibility model). In 1996, we established CH12F3 murine B lymphoma cells which can
switch from IgM to IgA with high efficiency in response to cytokine stimulation. Using this cell line we have shown that CSR breakpoints distribute in not only S
region but also its flanking regions which correspond to the intronic sequence for germline transcripts, suggesting correlation with germline transcription and CSR.
In 1998, we reported artificial substrates of CSR that undergo recombination in a cytokine-dependent manner when introduced into CH12F3 cells. Studies on such
substrates suggested that CSR recombinase recognizes not the primary sequence of S regions but their secondary structure. This hypothesis is further supported by our
recent study showing that stem-loop structure of DNA may be targeted by the CSR recombinase. We have also shown that the opposite orientation of S region
transcription (germline transcription) makes frequent inversion-type CSR in addition to deletion type. In addition, we found that the recombination efficiency is
positively correlated with transcriptional activity, suggesting association between recombination and transcription machineries. We speculate that transcription
transiently renders DNA single-stranded and induces formation of the stem loop structure in the S region containing abundant palindromic sequences. Such secondary
structure is likely to be recognized by CSR recombinase.
in 1978, (b) identification and characterization of S region as target of CSR in 1980, (c) molecular cloning
and physical mapping of the constant region genes, confirming their order on mouse chromosome predicted by the deletion model in 1982, (d) cloning of IL-4 and
IL-5 involved in regulation of CSR in 1986, and (e) demonstration of circular DNA byproducts excised during CSR in 1990. In 1986, Stavenezer and Alt found that
cytokine stimulation induces transcription from specific intronic promoters located 5' to each S region (germline transcription) and proposed that such transcription
makes recombination target S region accessible to a putative CSR recombinase (accessibility model). In 1996, we established CH12F3 murine B lymphoma cells which can
switch from IgM to IgA with high efficiency in response to cytokine stimulation. Using this cell line we have shown that CSR breakpoints distribute in not only S
region but also its flanking regions which correspond to the intronic sequence for germline transcripts, suggesting correlation with germline transcription and CSR.
In 1998, we reported artificial substrates of CSR that undergo recombination in a cytokine-dependent manner when introduced into CH12F3 cells. Studies on such
substrates suggested that CSR recombinase recognizes not the primary sequence of S regions but their secondary structure. This hypothesis is further supported by our
recent study showing that stem-loop structure of DNA may be targeted by the CSR recombinase. We have also shown that the opposite orientation of S region
transcription (germline transcription) makes frequent inversion-type CSR in addition to deletion type. In addition, we found that the recombination efficiency is
positively correlated with transcriptional activity, suggesting association between recombination and transcription machineries. We speculate that transcription
transiently renders DNA single-stranded and induces formation of the stem loop structure in the S region containing abundant palindromic sequences. Such secondary
structure is likely to be recognized by CSR recombinase.
Since we have found that the recombinase activity is induced de novo in CH12F3 cells upon cytokine stimulation, we carried out cDNA subtraction studies to
isolate genes whose expression is upregulated by CSR-inducing stimulation and found activation-induced cytidine deaminase (AID) which is specifically expressed in B
lymphocytes in germinal centers of lymphoid organs, the site of class switching and somatic hypermutation (SHM). The structure of AID (198 residues) has similarity to
that of APOBEC-1, a catalytic component of an RNA-editing enzyme complex required for cholesterol metabolism. Subsequent gene disruption experiments of AID revealed
indispensable roles of AID for not only CSR but also SHM. The same conclusion was drawn from a study of immune-deficient patients with hyper IgM syndrome type II,
in the AID gene of which we identified mutations. Since germline transcription of S regions and the general DNA repair machinery (NHES) are normal in
AID-deficient mice, AID is probably involved in the cleavage of DNA in CSR and SHM. Later, other group found AID is also essential for gene conversion (GC)
in chicken DT40 cells. Therefore, AID is an essential gene for all three genetic alteration systems of immunoglobulin gene that are driven by antigen stimulation.
The molecular mechanism how AID regulates CSR, SHM and GC is a puzzling and fascinating question to scientists in broad disciplines. Although these DNA
alterations are distinct in the overall mechanism and properties of the products, the initiation step of the three reactions is similar, namely all depend on the DNA
cleavage.
In 2001, the direct evidence to support involvement of AID in the DNA cleavage rather than the repair was obtained by experiments using focus formation
of a phosphorylated form of histone H2AX (g-H2AX) in the IgH locus as a marker of DSB (Petersen et al., 2001). Cleaved ends of DNA may be monitored by ATM/ATR or
DNA-PKcs, which phosphorylates histone H2AX, necessary for protection of DNA ends and for recruitment of many repair enzymes. Since H2AX deficient mice have severely
reduced CSR, g-H2AX focus formation is probably an important intermediate event of CSR. g-H2AX focus formation is induced at the IgH locus of spleen B cells upon
class switch stimulation. However, such focus formation as well as CSR is defective in AID deficient B cells stimulated for class switching, which indicates that
DSB in the IgH locus is dependent on the AID protein.
In 2002, we found that ectopic expression of AID induces class switching and hypermutation in non-B cells, such as fibroblasts and T cells, which carry
artificial constructs for measuring CSR and SHM. These results clearly demonstrate that AID is essential and sufficient to two different genetic alterations induced by
antigen stimulation of mammalian B cells. The other enzymes and cofactors are probably expressed ubiquitously. Transgenic mice with AID cDNA under the control of
ubiquitous promoter develop T cell tumors and die by 85 weeks without an exception. The onset of tumors varies from 4 to 40 weeks, depending on the copy numbers and
integration site of the transgene. In those T lymphomas, a large number of mutations accumulate in the V gene (10^-3), but very infrequently, in the C gene (10^-4)
of the T cell receptor, with a distribution profile of mutations reminiscent of SHM accumulation in Ig V genes. The mutation accumulation is also identified in
the c-myc gene. Mutation target genes are selective because there are many transcriptionally active genes, which do not have mutations. Therefore, mutation accumulation
leading to tumor development is not solely related with deficiency in DNA repair genes, but also to an uncontrolled AID activity, thus identifying AID as the
first active mutator in vertebrates.
Currently, the mechanism by which AID induces CSR is debatable. Two major hypotheses have been proposed, namely DNA deamination hypothesis and RNA
editing hypothesis. To determine which hypothesis is more valid, we performed series of experiments. In 2003-2004, we showed the requirement of de novo protein
synthesis after AID activation to induce CSR. And further study confirmed that the blockade occurs before the step of DNA cleavage. Thus, initiation of CSR should
require a newly synthesized protein after AID induction that is consistent with RNA editing hypothesis. In contrast, the inhibition of Uracil-DNA-glycosylase (UNG),
which is supposed to have major role in cleaving DNA in DNA deamination hypothesis, showed almost no effect on the formation of DNA double strand breaks. Besides,
we found that Ung mutants whose U removal activity is virtually null still could rescue CSR in ung knock out B lymphocytes. Taken together, we conclude that RNA
editing hypothesis is more likely than DNA deamination hypothesis.
Goal of Our Study
1) Isolation of the physiological target for AID2) Isolate of associating co-factors which work for CSR and/or SHM
3) Understanding the whole molecular mechanism of AID-induced immunoglobulin-gene diversification
Selected Publications
1. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the hyper-IgM syndrome (HIGM2).Revy, P., Muto, T., Levy, Y., Geissmann, F., Plebani, A., Sanal, O., Catalan, N., Forveille, M., D.-Lagelouse, R., Gennery, A., Tezcan, I., Ersoy, F., Kayserili, H., Ugazio, A.G., Brousse, N., Muramatsu, M., Notarangelo, L.D., Kinoshita, K., Honjo, T., Fischer, A. and Durandy, A.
Cell 102 565-575 (2000) [PubMed]
2. In situ class switching and differentiation to IgA-producing cells in the gut lamina propria.
Fagarasan, S., Kinoshita, K., Muramatsu, M., Ikuta, K. and Honjo, T.
Nature 413 639-643 (2001) [PubMed]
3. Constitutive expression of AID leads to tumorigenesis.
Okazaki, I., Hiai, H., Kakazu, N., Yamada, S., Muramatsu, M., Kinoshita, K. and Honjo, T.
J. Exp. Med. 197 1173-1181 (2003) [PubMed]
4. AID mutant analyses indicate requirement for class-switch-specific cofactors.
Ta, V-T., Nagaoka, H., Catalan, N., Durandy, A., Fischer, A., Imai, K., Nonoyama, S., Tashiro, J., Ikegawa, M., Ito, S., Kinoshita, K., Muramatsu, M. and Honjo, T.
Nature Immunol. 4 843-848 (2003) [PubMed]
5. Uracil DNA glycosylase activity is dispensable for immunoglobulin class switch.
Begum, N. A., Kinoshita, K., Kakazu, N., Muramatsu, M., Nagaoka, H., Shinkura, R., Biniszkiewicz, D., Boyer, L. A., Jaenisch, R. and Honjo, T.
Science 305 1160-1163 (2004) [PubMed]
6. AID-induced decrease in topoisomerase 1 induces DNA structural alteration and DNA cleavage for class switch recombination.
Kobayashi M, Aida M, Nagaoka H, Begum NA, Kitawaki Y, Nakata M, Stanlie A, Doi T, Kato L, Okazaki IM, Shinkura R, Muramatsu M, Kinoshita K, Honjo T.
Proc Natl Acad Sci U S A. 106:22375-80 (2009) [PubMed]
7. Histone3 lysine4 trimethylation regulated by the facilitates chromatin transcription complex is critical for DNA cleavage in class switch recombination.
Stanlie A, Aida M, Muramatsu M, Honjo T, Begum NA.
Proc Natl Acad Sci U S A. 107:22190-5 (2010) [PubMed]
8. Mice carrying a knock-in mutation of Aicda resulting in a defect in somatic hypermutation have impaired gut homeostasis and compromised mucosal defense.
Wei M, Shinkura R, Doi Y, Maruya M, Fagarasan S, Honjo T.
Nat Immunol. 12:264-70 (2011) [PubMed]
9. The AID dilemma: infection, or cancer?
Honjo T, Kobayashi M, Begum N, Kotani A, Sabouri S, Nagaoka H.
Adv Cancer Res. 113:1-44 (2012) [PubMed]
10. Nonimmunoglobulin target loci of activation-induced cytidine deaminase (AID) share unique features with immunoglobulin genes.
Kato L, Begum NA, Burroughs AM, Doi T, Kawai J, Daub CO, Kawaguchi T, Matsuda F, Hayashizaki Y, Honjo T.
Proc Natl Acad Sci U S A. 109 :2479-84 (2012) [PubMed]
11. Chromatin reader Brd4 functions in Ig class switching as a repair complex adaptor of nonhomologous end-joining.
Stanlie A, Yousif AS, Akiyama H, Honjo T, Begum NA.
Mol Cell 55:97-110 (2014) [PubMed]
12. APE1 is dispensable for S-region cleavage but required for its repair in class switch recombination.
Xu, J., Husain, A., Hu, W., Honjo, T. and Kobayashi, M.
Proc. Natl. Acad. Sci. USA 111 17242-17247 (2014) [PubMed]
13. Identification of DNA cleavage- and recombination-specific hnRNP cofactors for activation-induced cytidine deaminase.
Hu W, Begum NA, Mondal S, Stanlie A, Honjo T.
Proc Natl Acad Sci U S A. 112:5791-6 (2015) [PubMed]